2 TL ; DR - Guide d’évaluation des comparaisons à un groupe contrôle externe
3 Les études de comparaison externe, de quoi s’agit-il ?
5 Les problématiques méthodologiques soulevées par les comparaisons externes
6 Les comparaisons externes sont des études observationnelles
7 Position des agences de régulation et de HTA
§ FDA, USA
7.2 Utilisation, évaluation par les agences
8 De la nécessité d’avoir des preuves de l’intérêt cliniques des nouveaux traitements
9 Les sources de données utilisables
10 Les problématiques liées à l’aspect rétrospectif de ces études
12 Démarche hypothético déductive
13 L’inférence causale et les hypothèses sous-jacentes
15 Les techniques d’analyses statistiques
16 Le diagnostic d’absence de biais de confusion résiduel
18 Identifications des patients dans la source de données
21 Les outils d’évaluation du risque de biais
22 L’émulation d’un essai cible
23 Le benchmarking et les contrôles positifs
24 Analyses de sensibilité , analyses quantitatives du biais
26 Contrôle du risque alpha global
Les comparaisons externes sont explicitement mentionnées dans les utilisations reconnues par la FDA (Food and Drug Administration) des real world evidence envisageables pour informer les prises de décision et la construction des stratégies thérapeutiques (cf. Tableau 5) [4] . Elles sont classées dans les « non randomized interventional study » compte tenu de l’aspect expérimental (interventionnel) du groupe traité et dénommée « externally controlled trial ».
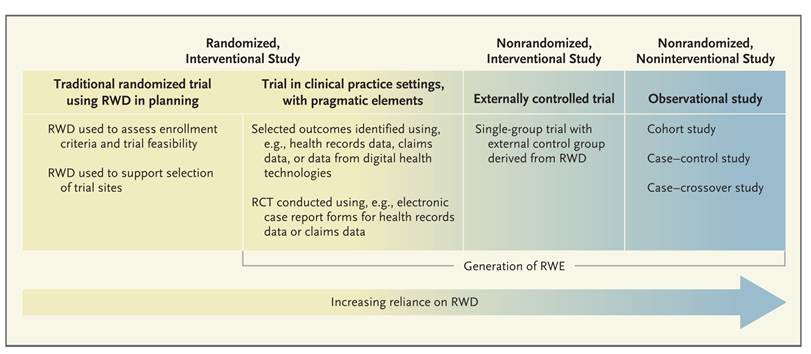
Tableau 5 – Utilisation envisagée des RWE par la FDA [4]
Les études de comparaisons externes se distinguent des études observationnelles où les deux groupes comparés sont issus des données de vraie vie (« nonrandomized, non-interventional study »).
Globalement la position de la FDA apparait plutôt précautionneuse n’envisageant la possibilité du recours à des comparaisons externes que si la réalisation d’un essai randomisé est impossible. Les guides fixent un haut standard pour la qualité des données, la transparence et le contrôle du biais de confusion (correspondant à ce qui est développé dans ce document) [43] [47] . Leurs attentes en termes de solidité de la démonstration restent identiques quelques soit le design de l’étude interventionnelle (essais clinique) ou non interventionnelle (études observationnelles) réf. [45] page 4.
Aucune guideline spécifique des comparaisons à un groupe contrôle externe n’a été publiée à la date de rédaction de ce document (décembre 2025). Le plus proche est celui sur les essais contrôlés à comparateur externe (externally controlled trials ) de la FDA qui concerne bien une comparaison à un groupe externe, mais prévue d’emblée avec l’étude monobras du nouveau produit [6] .
Cependant les problématiques méthodologiques posées par ces études et leurs solutions sont identiques à celle des études observationnelles inférentielles du bénéfice du traitement (comme les études de comparatives effectiveness). En l’occurrence les guidelines de ces études s’appliquent aux comparaisons à des groupes contrôles externes comme le récent guide FDA [48] .
Les recommandations concernant le recueil des données de vraie vie (registre, bases administratives, dossier médicaux informatisé, etc.) s’appliquent aussi au recueil des données pour le bras contrôle externe [49] [50] [51] [52] .
Tableau 6 – Liste des guides FDA traitant des problématiques rencontrées dans les comparaisons externes (entres autres)
Real-World Evidence: Considerations Regarding Non-Interventional Studies for Drug and Biological Products [53] |
Considerations for the Design and Conduct of Externally Controlled Trials for Drug and Biological Products [6] |
Considerations for the use of real-world data and real-world evidence to support regulatory decision-making for drugs and biological products [45] |
Real-World Data: Assessing Electronic Health Records and Medical Claims Data to Support Regulatory Decision-Making for Drug and Biological Products [51] |
Real-World Data: Assessing Registries to Support Regulatory Decision-Making for Drug and Biological Products [52] |
Plusieurs points méthodologiques fondamentaux sont explicitement précisés dans le document chapeau/générique « Considerations for the use of real-world data and real-world evidence to support regulatory decision-making for drugs and biological products [45] » publié en aout 2023 :
Fin 2025, la FDA a lancé un programme pour 2026 d’incitation à la recherche dans la méthodologie de l’évaluation et de la régulation (regulatory science ) note n° 10 .
[10] https://sam.gov/workspace/contract/opp/5f582391365645c2b030260aaf922e8b/view.
